
Late-stage attrition kills returns on pharma R&D investment. One way to reduce attrition is to take less risk – incremental innovation reduces the risk of failing in development, but increases the risk of failing in the marketplace with even bigger (though delayed and less sudden) consequences.
The other solution is to address the principle causes of late stage failure: inadequate target validation and unpredictable safety liabilities.
Easier said than done? Not any longer – there is a single tool that can address both problems in one experiment. That tool is the Phenome-wide Association Study, or PheWAS, and it’s already delivering a quiet revolution across the industry.
Chances are, though, unless you work for one of the handful of global pharma that have been able to embrace this technology, you have haven’t even heard of PheWAS, or certainly appreciated what it can do for drug discovery and development in the 21st Century.
The reason for its stealth is simple enough: constructing a PheWAS platform not only consumes millions of dollars, but takes a decade or more to deliver – so only those with the right combination of cash and foresight are now sitting on the one tool that can deliver a step-change in Phase 3 success rates.
As the name suggests, PheWAS studies are close cousins of the better-known GWAS (Genome-wide Association Studies). You know the drill: take a few thousand people with a particular phenotype (often, but not always, a disease) and capture a detailed snapshot of their genomes, encoded in a million or more single nucleotide polymorphisms selected for maximal information density, and compare them to a few thousand healthy people lacking the phenotype under study. Clever statistics trawls the resulting vast databank to identify genetic variations associated with the phenotype.
A decade ago, GWAS was touted as the “next big thing”, the way of converting the hard-won human genome sequence into real understanding about real diseases – understanding that could drive the development of new treatments for common, but poorly understood, diseases.
The reality, though, has been much more underwhelming. At least one, and more often, several GWAS studies have been published for every major disease and the results have been interesting in a very unexpected way. Yes, such studies typically identify genes (or at least genetic regions) that are significantly associated with the phenotype, but the surprising finding is not the identity of the genes but the small fraction of population-wide risk of inheriting the phenotype they actually explain.
Such a conclusion is typically hidden by an abstract that pronounces a new list if candidate genes associated with a given complex trait. But look behind the qualitative listing at the quantitative assessment of association and the percentage of total variability explained by the identified genetic traits is usually tiny – single digit percentage points in aggregate for all the best markers identified across the whole genome.
What that means in practice is that none of the identified candidate genes are associated strongly enough with the phenotype to make them good candidates as therapeutic targets. More than a decade later, there are no approved products or late-stage clinical development projects that can claim their genesis in a GWAS dataset.
All this is in stark contrast to the success of human genetics in identifying targets for rare diseases. We have witnessed a golden-age of rare disease drug discovery and approvals, and though arguments may smolder on about the economics of high-priced orphan drugs, few dispute the successful exploitation of multiple new targets in this space.
The distinction is simple: genetic approaches have proved enormously successful at identifying and validating targets in diseases where one, or a small number, of genes (often encoding proteins involved in the same pathway) are involved. By contrast, in diseases where many genes are involved – which includes all the prevalent diseases of middle and old age that contribute the majority of unmet medical need at least in the developed world – the diseases where GWAS was meant to be the panacea – genetic approaches have delivered few if any truly useful insights.
PheWAS turns GWAS on its head
GWAS attempts to select among many genetic variants the few that are associated with a single, particular phenotype. PheWAS attempts to select among many phenotypes (the ‘phenome’ being the collection of all phenotypic characteristics of an individual) the few that are associated with a single, chosen gene.
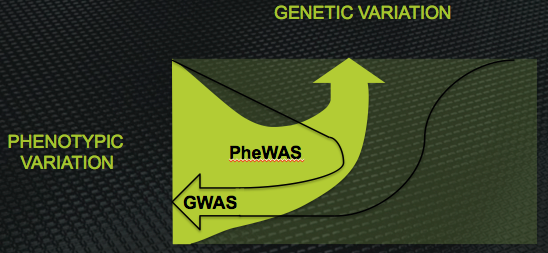
Diagramtic representation of the inverse relationship between the design of GWAS and PheWAS. GWAS links many genes to one phenotype; PheWAS links many phenotypes to one gene
This immediately delivers a couple of advantages. First, you can define phenotypes in multiple different ways without having to run the whole study again. In a GWAS, the definition of the phenotype is used to select the subjects included in the study, so if you change your mind even subtly about the phenotype of interest, the only solution is to go back and start again. In PheWAS, multiple related phenotypic definitions of similar conditions can be included in a single analysis.
This matters a lot for many highly prevalent degenerative diseases. They are often highly heterogeneous, with different subgroups having potentially very different causes. Many are still based on diagnostic criteria defined a century or more ago, which potentially encompass phenotypes that present very similarly but have no similarity in molecular origin. This heterogeneity is one often cited reason why GWAS studies usually disappoint when applied to such diseases: if the phenotype used to select the subject actually includes, say, four different diseases with four different pathogenic mechanisms, the statistical power of the study just melts away.
The second advantage is that by focusing on just one gene it is possible to capture essentially all the variability in that one gene. In contrast, even with modern genotyping technology, GWAS achieves only thin coverage of the whole genome. The problem is not (any more) the cost of acquiring more data, but an unavoidable consequence of statistics. The more genetic variants that are included in the analysis, the more subjects need to be entered into the study. There is, therefore, a necessary trade-off between study size and fine-scale detail.
Once upon a time, it was assumed (hoped?) this would not matter. If a single variant in a gene was principally responsible for the phenotype, local linkage would transmit that signal into “marker” haplotypes in the broad vicinity of the causative variant.
But experience with rare disease genetics, where one (or a handful) of genes are involved, usually identified through family studies, has amply illustrated that several, and often many, variants in the gene can cause essentially the same phenotype. If you have dozens of causative variants in the same gene contributing to the disease, then in the absence of fine detail in the genome “fingerprint” you will struggle identify the candidate gene at all (even if it is, really, strongly associated).
PheWAS, then, is much more powerful than GWAS in the real world we live in
But alongside the two advantages, come two disadvantages that must be overcome before you can begin to harvest its power.
The first and most obvious is that high-density genotyping has been industrialized, but high-density phenotyping is still in its infancy and most, if not all, of it still has to be done manually, one phenotype and one subject at a time. Assembling, then, a moderately deeply phenotyped cohort is an expensive and time-consuming activity, orders of magnitude greater in scope than implementing a G WAS. Counterbalancing that, though, is the advantage that you only ever need to do it once since the subjects included in such a cohort need not be selected at all.
The second disadvantage is that you have to start with a gene you care about, rather than a phenotype you care about. GWAS feels “unbiased” and a hypothesis generator – although it does have a hypothesis at its core (namely, that some gene(s) are associated with disease X as defined by the selected phenotype). PheWAS by contrast has as its core hypothesis “Gene X is interesting, what does it do?”
That’s not as big a disadvantage is it sounds. Biology is constantly throwing up enticing targets for candidate therapeutic interventions. In vitro studies, reverse genetics (knock-out animal models), and increasingly in silico network modeling all yield hundreds of plausible candidates each year. The problem for VCs, biotech companies and pharma companies alike is validating those targets.
That problem is compounded by the difficulty of even selecting the right indication to pursue. Once a knockout study (for example) has identified as a gene as yielding an interesting phenotype in mice, deciding how that may translate into human disease is a perilous extrapolation. For a start, the initial researchers likely only observed a small part of the knockout phenotype (either the most obvious part, or the part that overlapped with their pre-existing interests). Potentially the component of the phenotype most relevant to human disease may have been overlooked altogether.
PheWAS provides a straight-forward solution. If any of the plethora of academic biology experiments suggests a gene is potentially interesting, then a PheWAS study will tell you which phenotypes in the human population are most strongly associated with the function of that gene.
A decade ago, GWAS was touted as the “next big thing”, the way of converting the hard-won human genome sequence into real understanding about real diseases. The reality, though, has been underwhelming
In some sense, PheWAS is like performing a knockout study in man without the impossible ethical hurdle. And its much more subtle – knockouts typically only reveal the consequences of loss of function of the gene product, usually in all tissues for the whole life of the organism. PheWAS, on the other hand, reveals the phenotypic consequences of much more subtle gain or loss of function, perhaps in certain tissues or at certain times in the lifecycle of the organism (because the myriad genetic variations in the population each confer subtly different functional consequences).
If the most obvious application of the ability of PheWAS to tell you which disease phenotype is most strongly linked to modulation of the activity of a particular gene product lies in target validation, its most valuable application arguably lies in predicting side-effects of modulating a new target.
With a wide range of ‘intermediate phenotypes’ included in the PheWAS design, the output provides links not only between the gene of interest and global physiological phenotypes, such as disease, but also between the gene of interest and particular molecular pathways.
This can (and does) reveal unexpected associations with apparently unrelated pathways. Most usefully, that allows specific safety investigations to be included in small, early and above all low cost, clinical trials rather than waiting for the toxicity to reveal itself only in later, larger trials. Today, those late-stage safety failures, when the toxicity emerges from left-field are just branded ‘unlucky’. But de-railing flagship first-in-class phase 3 programmes costing tens or hundreds of millions, they have a massive impact on the overall productivity of pharma R&D. Tomorrow, PheWAS can eliminate these surprising toxicities associated with previously untested targets – though, of course, genuine off-target toxicities associated with a given molecule will always remain a threat.
So much for the theory, then. How does PheWAS work in practice?
The first requirement is a large cohort of deeply-phenotyped subjects from whom DNA is available for analysis. ‘Large’ in the context means a few thousand; from a statistical perspective, the number of people need not be as large as a typical GWAS because (at least today) the phenotyping density is lower than a million-SNP genotyping panel. For PheWAS the major consideration is having enough representatives of rarer phenotypes within a broadly unselected population. Accessing phenotypes with a population frequency above 1% is relatively accessible with a few thousand subjects enrolled.
The deeper these subjects are phenotyped, the more useful will be the output. Our MaGiCAD cohort, which is one of the few resources for PheWAS studies available on a fee-for-service basis, has about 6,000 phenotypes defined, ranging from diseases (coronary heart disease, arthritis, depression, overactive thyroid, overactive bladder and many others), through specific molecular measures (total cholesterol, triglycerides, glucose) to high-density multi-marker panels (multiplexed cytokine data, metabolic profile, immunological profile).
Having selected the gene of interest, a haplotype analysis is then performed to identify the minimum set of marker SNPs that will capture >98% of the genetic variation at that locus. There seems little point capturing rarer variants, since there is only power to capture phenotypes with at least 1% prevalence in any case. Typically, this involves anywhere between three and twelve SNPs.
These SNPs are then determined for all the subjects in the cohort (a rapid, low-cost activity with today’s high throughput, accurate genotyping tools).
The final, and most demanding, stage is the statistical analysis. Since the design of a PheWAS study is just the logical inverse of a GWAS study, the general approaches are well defined. However, there is one key difference: the correlation structure of a genomic fingerprint is defined by linkage (that is, SNPs that are physically closer to one another lying on the same DNA molecule are more likely to be correlated). This well-understood linkage provides a useful baseline for the statistical analysis, and increases the power considerably compared to treating each SNP as an unrelated data point.
While the need for such ‘tricks’ is lessened by the shallower data density in a phenotyped cohort (MaGiCAD has 6,000 phenotypes; a GWAS study typically has a million or more SNPs from each subject, so the data density is two orders of magnitude lower in the PheWAS design, at least with today’s phenotyping capabilities), there is nevertheless a correlation structure within the ‘phenome’ too. In other words, certain phenotypes are much more likely to occur with each other (for example, coronary heart disease and type 2 diabetes). This allows something akin to a ‘linkage map’ for phenotypes to be constructed, and used to turbo-boost the statistical power in PheWAS.
And what does the output look like in a real-world example?
One of our companies, Epsilon-3 Bio, is studying compounds that modulate the functions of a novel target, apolipoprotein E (or apoE). ApoE has a well-known, and well understood, role in lipid metabolism, and is also known to be associated with Alzheimer’s Disease (although the molecular mechanism behind that association is much less clear). Are these the best, or only, diseases that might be amenable to treatment with apoE modulators?
Epsilon-3 Bio engaged Total Scientific to run a PheWAS study with apoE at the “candidate gene”. The results were impressive: in addition to a strong signal highlighting the already-known associations (providing a useful validation of the technique), the data highlighted a powerful, and novel, link between apoE genotype and major depression. Simultaneously, this provides insight into the pathogenesis of clinical depression, supporting the idea that changes in brain architecture may be the substrate for depressive symptoms, and suggests a new application of our apoE modulators – as the first disease-modifying therapeutics (as opposed to symptomatic treatments) in the huge depression market.
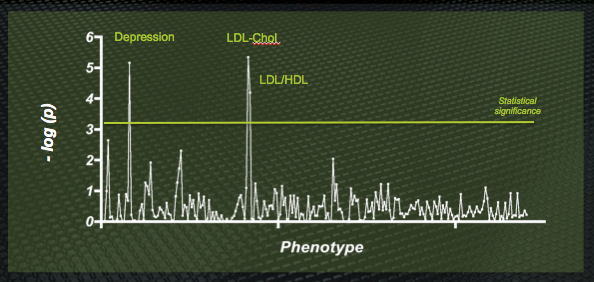
Sample output from a PheWAS study of the apoE gene. The well-known association with LDL cholesterol levels (since apoE is the ligand for the LDL receptor) is found – but a previously unexpected association with major depression is also highly significant.
Separately, the PheWAS data highlighted some novel molecular pathways that may be associated with altered apoE activity, yielding a couple of additional analysis that will be added to early stage clinical trials of any apoE modulators, reducing the risk of a late-stage failure due to unexpected on-target effects.
PheWAS, then, is delivering actionable answers to the most pressing questions in the development of high-value first-in-class therapeutics. Selecting target indications based on human biology rather than preclinical models will principally improve outcomes in Phase 2a proof-of-clinical-concept studies, but may also improve Phase 3 success rates in those indications where earlier proof of principle is impossible or unreliable. Knowing which pathways are likely to deliver mechanism-based toxicities and side-effects will allow targeted investigations in early stage development, and reduce late stage attrition accordingly.
There are less than a hundred PheWAS studies in the scientific literature today – and many of those have only a handful of phenotypes included (sometimes as few as a dozen). However, that reflects who has access to the technology in practice, rather than its limited utility. At Index Ventures, performing a PheWAS study at the very beginning of an asset-centric development programme for unvalidated targets has already become the default path. Wider adoption will surely improve pharma R&D metrics.
The next five years will see a steady increase in the application of this valuable tool. Only the limited availability of publically-accessible PheWAS resources (together with the huge time barrier to creating new ones) will prevent an explosion in use. Those interested in improving their return on R&D investment should form an orderly queue.
DrugBaron is Chairman of Total Scientific Ltd, the global leader in fee-for-service PheWAS studies.
Pingback: Next Hurdle For Medical Research: Capture And Integration Of Phenotype At Scale | Next Hurdle For Medical Research: Capture And Integration Of Phenotype At Scale | Social Dashboard()
Pingback: Next Hurdle For Medical Research: Capture And Integration Of Phenotype At Scale | zipicon()